Генетические основы гемобластозов
Вступление
Изучение патогенеза опухолевых заболеваний системы крови привело к открытию одного из ведущих принципов онкогенеза в целом: причиной формирования злокачественного клона является нарушение функционирования нормальных генов. Как правило, это происходит в результате хромосомных аберраций, мутаций отдельных генов или блокирования нормальной регуляции функционирования генов в связи с эпигеномными (не связанными непосредственно с повреждением структуры генов) событиями.
Молекулярная генетика гемобластозов как отдельное научное направление стала развиваться в связи с успехами цитогенетики, которая, начавшись как дисциплина описательная, в дальнейшем позволила успешно идентифицировать гены, участвующие в хромосомных нарушениях.
В последние годы во всем мире продолжается активная исследовательская работа по идентификации генетических аномалий при гемобластозах и изучению их влияния на процессы жизнедеятельности клетки. Известно большое количество генетических нарушений, ведущих к развитию определенных видов опухолевых заболеваний крови. Детекция аномальных генов в настоящее время является обязательным условием для установления диагноза целого ряда гемобластозов.
Более того, выявление определенных транслокаций и мутаций позволяет достаточно уверенно предполагать особенности течения заболевания, судить в ряде случаев о прогнозе и подбирать адекватную терапию. Особое значение приобрело развитие основанной на полученных знаниях так называемой «прицельной терапии», позволяющее проводить коррекцию имеющихся нарушений на генетическом или биохимическом уровне.
Самыми яркими примерами лекарственных препаратов, применяемых как «прицельная терапия», являются полностью трансретиноевая кислота (АТРА) и иматиниба мезилат (Гливек), позволившие внести революционные изменения в терапию острого промиелоцитарного лейкоза (ОПЛ) и хронического миелолейкоза (ХМЛ).
Общие представления
Популярное ранее представление, что участие генов в транслокациях, нарушающих структуру их ДНК, само по себе придает им способность трансформировать клетку, сейчас кажется несколько упрощенным. Традиционно гены, участвующие в канцерогенезе классифицировались в одну из двух категорий: протоонкогены или опухолевые супрессоры. В настоящее время выделена третья категория: гены, поддерживающие целостность ДНК или обеспечивающие реконструкцию ДНК вследствие повреждений. Участвуя в аберрациях, эти гены не обладают способностью к трансформации, однако способствую накоплению трансформирующих мутаций.
Таким образом, эти три категории генов при любом виде повреждения прямым или непрямым способом ведут к малигнизации клетки.
Онкогены: исторически, гены, захваченные быстро трансформирующими ретровирусами из генома клетки называюся онкогенами, а их клеточные гомологи – протоонкогенами. В более широком смысле, онкогенами называются любые гены, образование доминантных мутаций в которых ведет к малигнизации клетки.
В настоящее время известно боле 30 ретровирусных онкогенов, имеющих клеточные аналоги. Лишь небольшая часть из них участвует в транслокациях, приводящих к развитию гемобластозов, но практически все задействованы в основных регуляторных путях. Гены, вовлеченные в транслокации, могут вызывать трансформацию клетки с помощью двух механизмов. Классическим примером первого из них является транслокация t(9;22), когда происходит формирование химерного онкогена bcr/abl, белок которого – тирозинкиназа – обладает непосредственным трансформирующим потенциалом.
Подобное происходит а транслокациях, вовлекающих гены MLL, RUNX1, RAR. Второй механизм активации часто встречается при лимфоидных опухолях, когда транслокация затрагивает гены тяжелых цепей иммуноглобулинов или Т-клеточных рецепторов. При этом второй ген-участник транслокации, в норме «молчащий» на этой фазе клеточной дифференцировки, начинает усиленно экспрессироваться. Это обычно приводит к остановке дифференцировки клетки на определенной стадии и усиленной пролиферации.
Гены-супрессоры опухоли: Обнаружение хромосомных делеций, часто возникающих при определенных видах опухолей, привело к выявлению генов-супрессоров опухоли, потеря которых вызывает злокачественную трансформацию клетки. Подразумевается, что при потере одного гена одновременно должна происходить механическая или функциональная потеря такого же гена на второй хромосоме, для того, чтобы реализовался негативный эффект мутации.
Этот механизм часто задействован при солидных опухолях (например, RB1 при ретинобластоме, TP53 при опухолях толстого кишечника). Известно около 30 регионов, расположенных на 13 хромосомах, в которых регулярно происходят делеции или обнаруживается потеря гетерозиготности, ведущие к развитию онкогематологических заболеваний.
Как ни странно, лишь в некоторых из них обнаруживаются гены, претендующие на название опухолевых супрессоров. В настоящее время рассматривается гипотеза, что в ряде случаев бывает достаточно мутации лишь одной аллели гена-супрессора для реализации трансформирующего эффекта. Предположительно, значение имеет так называемая «доза» гена.
Гены, поддерживающие целостность ДНК. Мутации в этих генах не приводят непосредственно к развитию опухолей, однако, в результате возрастания генетической нестабильности, способствуют накоплению мутаций в клетке. Зачастую эти гены, относящиеся к микросателлитам (например, MLH1 и MSH2) мутируют в случаях солидных опухолей. Для гемобластозов такие виды мутаций нехарактерны.
Отражением другого вида генетической нестабильности являются множественные хромосомные аберрации у пациентов ОМЛ и МДС из группы высокого риска. Как правило, течение таких видов лейкемии сопровождается быстрым развитием резистентности к терапии или возникновением ранних рецидивов. Очевидно, наличие множественных хромосомных аберраций является отражением ранних событий в лейкемической клетке, скорее всего, повреждением механизма восстановления двухцепочечной ДНК. Известен ряд генов, которые, возможно, вовлечены в этот процесс (ATM, TP53, MRE11).
Генетические аномалии при острых миелоидных лейкозах
Основными молекулярными событиями, ведущими к формированию лейкемического клона при миелоидных лейкозах, являются либо возникновение специфических транслокаций, зачастую с вовлечением протоокогенов, либо мутации генов, участвующих в контроле пролиферации и дифференцировки миелоидной ткани. Для миелоидных опухолей наиболее характерными являются реципрокные транслокации, при которых происходит обмен генетическим материалом между различными хромосомами с образованием патологических хромосомных структур, самой известной из которых является Филадельфийская хромосома.
На молекулярном уровне в процессе такой транслокации образуется так называемый химерный ген, состоящий из активных участков (доменов) двух генов-участников перестройки и ведущий к экспрессии химерного белка, который способен, как правило, либо блокировать миелоидную дифференцировку, либо стимулировать бесконтрольную клеточную пролиферации за счет следующих событий:
- нарушение функционирования ядерных рецепторов (характерный пример – острый промиелоцитарный лейкоз);
- подавление транскрипции (считывания РНК с ДНК) за счет связывания ключевого белкового комплекса CBF (core binding factor), что характерно для острых лейкозов с транслокациями, вовлекающими 21 и 16 хромосомы (гены RUNX-1, бывший AML-1, и CBFB);
- подавление генов гомеобокса, или регуляторов клеточного развития (характерно для ОМЛ с транслокациями, в которых участвует ген MLL – mixed lineage leukemia-, расположенный на 11 хромосоме);
- бесконтрольная активация ферментов-тирозинкиназ (BCR-ABL позитивные миелоидные лейкозы, мутации гена FLT3);
Особо хотелось бы отметить важность мутаций генов, не вовлеченных в транслокации, но являющихся медиаторами процессов, описанных выше. К ним относятся частичная тандемная дупликация гена MLL в случае соматической мутации 11q23, внутренняя тандемная дупликация гена FLT3, ведущая к активации киназного каскада за счет маскировки под нормальный киназный рецептор, мутации генов СЕВРА и RUNX1 (AML1), подавляющих транскрипцию за счет связывания комплекса CBF, мутации гена NPМ, приводящие к блоку образования белков в рибосомах.
| Генетическая аномалия | Гены, участвующие в перестройке | Функция протеина | Частота встречаемости при ОМЛ |
|---|---|---|---|
| t(15;17) t(11;17) t(11;17) t(5;17) t(17;17) |
PML/RARA PLZF/RARA NuMa/RARA NPM/RARA STAT5b/RARA |
Рецептор ретиноевой кислоты | 95% М3 менее 5% М3 менее 1% М3 менее 1% М3 |
| t(8;21) t(3;21) t(6;21) |
AML1/ETO AML1/Evi1 AML1/MTG16 RUNX1 mut |
Транскрипционный фактор | 20% М2 менее 1% М2 и МДС менее 1% М2 и МДС 3-5% М2 без цитогенетически определяемых хромосомных аномалий |
| inv 16 t(16;16) |
CBFB/MYH11 | Транскрипционный фактор | Практически 100% М4 Ео |
| t(9;22) | BCR/ABL | Тирозинкиназа |
До 20% ОМЛ (у детей -5%, у взрослых – 15-25%) |
| t(6;11) t(9;11) t(10;11) t(11;19) 11q23 |
AF6/MLL AF9/MLL AF10/MLL MLL/ENL MLL ITD |
Транскрипционный фактор | До 15- 20% М4 и М5, включая суммарно все нарушения MLL До 11% ОМЛ без цитогенетически выявляемых хромосомных аномалий |
| t(3;5) | NPM/MLF1 | Регулятор трансляции | 1-2% всех ОМЛ |
| FLT3 ITD | Рецептор тирозинкиназы | до 30% при М3, также при ОМЛ без цитогенетически выявляемых хромосомных аномалий | |
| CEBPA mut | Транскрипционный фактор | 16% М2 без цитогенетических хромосомных аномалий | |
| NPM mut | Регулятор трансляции | 30% всех ОМЛ, до 60% ОМЛ без цитогенетически выявляемых хромосомных аномалий |
Значение химерного гена PML/RARA при остром промиелоцитарном лейкозе и его роль в блоке миелоидной дифференцировки
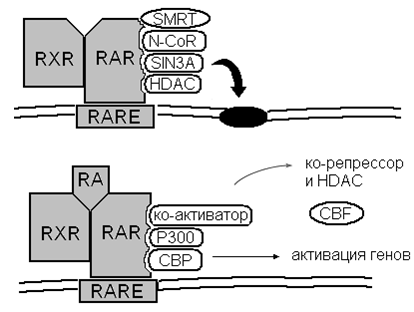
На рис. 3 изображена нормальная регуляция миелоидной дифференцировки в результате активации рецептора ретиноевой кислоты α (RARα). В неактивном виде рецептор образует подавляющий транскрипцию комплекс вместе с другим рецептором этого же семейства (RXR), ядерными помощниками – корепрессорами (SMRT, N-CoR, Sin3a) и белком гистондеацетилазой HDAC.
Этот комплекс соединяется со специфическим участком ДНК (RARE – retinoid acid responsive element), полностью блокируя считывание РНК. В нормальной ситуации рецептор связывается с ретиноевой кислотой (RA) и связь с корепрессорами разрушается. Далее, вместо корепрессоров, происходит присоединение коактиваторов (P300, CBP), что приводит к активации нормальной миелоидной дифференцировки.
При образовании транслокации t(15;17), ведущей к появлению химерного белка PML/RARA, возникает прочная связь рецептора с корепрессорами, не разрушающаяся в присутствии физиологических концентраций полностью транс-ретиноевой кислоты (ATRA). ДНК освобождается лишь в присутствии фармакологических концентраций ATRA, превышающих физиологическую в тысячи раз. Только это делает возможной нормальное функционирование белкового комплекса и дальнейшую миелоидную дифференцировку (10).
Тот же механизм работает и при транслокации t(5;17), вовлекающей гены NPM и RARA. Совершенно другая ситуация возникает при ОПЛ с транслокацией t(11;17) (PLZF/RARA) и t(11;17) STAT5b/RARA. Ген PLZF, так же как и STAT5b, имеет собственные зоны контакта с комплексом корепрессоров и добавление ретиноевой кислоты не приводит к освобождению зависимых генов для нормальной экспрессии. Сложность диагностики при остром промиелоцитарном лейкозе заключается в том, что морфологические и цитохимические методы не позволяют различать между собой разные виды характерных транслокаций.
Стандартная цитогенетика зачастую неэффективна, так как при ОПЛ клетки костного мозга в обычной краткосрочной культуре делятся плохо, и материала не хватает для подтверждения диагноза ОПЛ и дифференцирования транслокаций. Все вышеуказанные транслокации возможно определять с помощью флюоресцентной in situ гибридизации (FISH) или метода обратной транскриптазной реакции с проведением далее так называемой гнездной полимеразной цепной реакции.
Рис. 3. Нормальная регуляция миелоидной дифференцировки путем активации рецептора ретиноевой кислоты.
Таким образом, определение транслокаций при ОПЛ является крайне необходимым не только для установления правильного диагноза, но и для определения первичной резистентности к стандартной для ОПЛ терапии ATRA и, соответственно, к определению прогноза (благоприятного при t(15;17) и t(5;17) и неблагоприятного при всех остальных видах транслокаций).
Значение определения транслокаций, участвущих в репрессии комплекса CBF
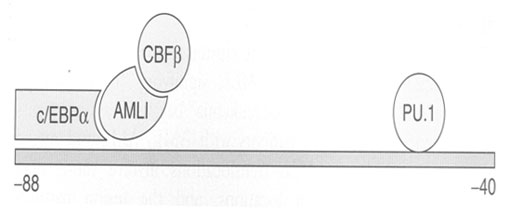
Образование нормального комплекса CBF, ведущего к активации гена макрофагального колониестимулирующего фактора (M-CSF), играет важнейшую роль в миелоидной дифференцировке и напрямую зависит от формирования связи между белками-продуктами генов c/EBPα, AML1(RUNX1) и CBFβ (рис 4). Поэтому, все транслокации, вовлекающие хотя бы один из перечисленных генов, такие как t(8;21) или inv 16 превращают указанный комплекс из активатора в репрессор, и таким образом блокируют миелоидную дифференцировку.
Блокада происходит с участием ядерных корепрессоров и гистондеацетилаз, тех же что были описаны в предыдущем разделе, касающемся лейкемогенеза при ОПЛ. Надо отметить, что любые мутации генов, участников комплекса CBF, таких как c/EBPα, RUNX1, даже при отсутствии цитогенетически определяемых нарушений ведут к формированию лейкемического фенотипа, то есть, нарушению дифференцировки и бесконтрольной пролиферации. Тем не менее, по современной классификации, обнаружение данных нарушений относят этот тип лейкоза в благоприятную группу (при условии проведения соответствующей терапии).
Рис. 4. Образование комплекса CBF
Обычно транслокации t(8;21) или inv 16 и др. довольно легко определяются при стандартном цитогенетическом исследовании. Так же, как и при ОПЛ, используется обратно-транскриптазная полимеразная цепная реакция. При определении мутаций генов c/EBPα, RUNX1 с наибольшей эффективностью может быть применена только ПЦР.
Если учесть, что данные аберрации суммарно встречаются примерно в 15-20% М2 без цитогенетически выявляемых нарушений, становится ясной необходимость выявления этих мутаций, тем более, что по последним данным, обнаружение таких аномалий без сочетания с другими цитогенетическими нарушениями относит этот тип лейкоза из группы промежуточного в группу благоприятного прогноза.
Определение транслокаций, ведущих к активации тирозинкиназ и киназных рецепторов.
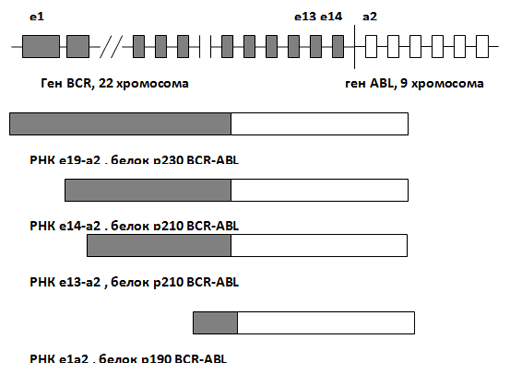
Самой известной транслокацией, приводящей к аномальной активации киназного каскада и неконтролируемой клеточной пролиферации, является Филадельфийская хромосома или t(9;22), ведущая к образованию химерного гена BCR/ABL (рис 5). В зависимости от локализации точки разрыва на 22 хромосоме образуется химерный онкоген различной длины (от 190 до 230 килодальтон).
Продукт этого гена (тирозинкиназа) ведет к фосфорилированию огромного количества протеинов (RAS, PI-3, Grb2, Crkl), участвующих в процессе сигнальной трансдукции – передаче сигнала с рецепторов клеточной мембраны ядерному генетическому материалу. Активация такого количества различных сигнальных путей ведет к независимой от ростовых факторов пролиферации, нарушению адгезии клеток к стромальному окружению и устойчивости к апоптозу – программированной клеточной гибели. При остром миелоидном лейкозе встречается как тип транслокации с точкой разрыва M-bcr, ведущий к образованию протеина размером 210 килодальтон (р210), так и m-bcr, с образованием протеина p190.
Рис. 5. Образование химерного гена BCR/ABL.
Другим вариантом самопроизвольной стимуляции киназного каскада является выявление мутации гена FLT3 (FMS-like tyrosine kinase receptor), кодирующего тирозин-киназный рецептор. Образование так называемой внутренней тандемной дупликации в структуре гена ведет к изменению третичной структуры рецептора таким образом, что он мимикрирует под обычный рецептор, уже активированный специфическим лигандом.
Это ведет к активации протеинкиназ ERK2 и AKT, приводит к неконтролируемой пролиферации и обычно ассоциируется с выраженным лейкоцитозом. Следует особо отметить, что выявление этого нарушения у пациентов с t(15;17) или у больных без явных нарушений кариотипа резко ухудшает прогноз и переводит этих пациентов в группу неблагоприятного течения ОМЛ.
По данным последних исследований, все виды нарушений, связанных с активацией киназных сигнальных путей являются независимым отрицательным прогностическим признаком.
Значение выявления транслокаций, ведущих к нарушению экспрессии генов гомеобокса
К генам гомеобокса относят целый ряд генов, кодирующих транскрипционные факторы, которые принимают участие в регуляции эмбрионального развития. Эти гены имеют высоко гомологичную структуру у большинства эукариот, от самых низших до наиболее высоко организованных. Наиболее значимым геном-регулятором экспрессии генов гомеобокса у млекопитающих является ген MLL (mixed lineage leukemia).
Партнерами этого гена являются гены-регуляторы транскрипции, поэтому любые мутации, вовлекающие MLL, ведут к подавлению транскрипции генов гомеобокса. Точный механизм лейкемогенеза в данном случае пока до конца не определен. MLL участвует в образовании более 25 различных транслокаций, которые (кроме t(4;11)) обычно характерны для ОМЛ М4/М5. Наиболее известным и распространенным нарушением является соматическая мутация 11q23, приводящая к образованию частичной внутренней тандемной дупликации, в которой участвуют экзоны со 2 по 6 или со 2 по 8. Подобное нарушение определяется в случаях трисомии 11 хромосомы и в 11-20% случаев ОМЛ без цитогенетических нарушений. Следует отметить, что любое нарушение структуры гена MLL однозначно определяет неблагоприятный вариант течения ОМЛ.
Мутации гена NPM
Ген нуклеофозмина (NPM-1) в последнее время привлекает все более пристальное внимание исследователей, ввиду частой выявляемости мутаций этого гена при ОМЛ с нормальным кариотипом (50%-60%). В норме продукт этого гена играет ключевую роль в образовании протеосом и, таким образом, опосредованно влияет на синтез белков в клетке. Ген NPM-1 участвует в ряде реципрокных транслокаций, характерных для анаплазированной Т-клеточной лимфомы t(2;5), редко встречающихся форм МДС с транслокацией t(3;5), и крайне редкого типа ОПЛ с t(5;17).
Для ОМЛ более характерны мутации NPM-1, наиболее частой из которых является дупликация тетрануклеотида TCTG в позиции 956-959 в 5 экзоне (75-80% случаев). Во всех случаях мутаций или транслокаций с участием NPM-1 происходит изменение нормального внутриядерного расположения белка на цитоплазматическое и нарушается его взаимодействие с генами – партнерами (ТР53 и ARF), что инактивирует их как опухолевые супрессоры. Тем не менее, обнаружение мутаций NPM-1, как правило, ассоциируется с благоприятным течением ОМЛ (за исключением случаев ассоциации с мутациями гена FLT-3). В настоящее время продолжается активное изучение этого гена с целью уточнения его роли в лейкемогенезе и возможного использования его как маркера при исследовании минимальной остаточной болезни.